
| What is the TACOS server? |
TACOS server is an internet service for prediction of protein complex structure prediction using a hybrid of threading, ab initio and protein-protein docking . While most efforts in the field of protein complex structure prediction is concentrated on protein-protein docking, it suffers from a severely limited in scope since docking can only be performed when the unbound protein structures are known before hand.TACOS aims to bridge this gap by predicting protein complex structures from the primary amino acid sequence by the aided of identification of protein complex template structures.
| How does TACOS identify the best templates? |
When the users submit a protein complex sequence, the TACOS server attempts to identify
a complex sequence from a protein complex structure library which is most homologous/
structually analogous to the submitted query sequence using the
COTH and SPRING algorithms.
COTH attempts to identify the
best template using a profile-profile alignment based approach where the scoring function
includes a number of predicted structural features to aid profile-profile alignment. The
scoring functions include 1) Match between the sequence profiles of the query and the template
2) Match between the predicted secondary structure of the query and the native secondary structure
of the template. 3) Match between the sequence profile of the query and a depth based structure
profile of the templte. 3) Match between the predicted solvent accessibility of the query residues and
the native solvent accessibility of the template residues. 4) Match between the predicted torsional
angles (phi and psi angles) of the query residues and the native torsional angles of the templat e residues.
5)Match between the hydrophcity of the query and template residues 6) Match between the predicted interface
residues of the query and the native interface residues of the template complex. In order to prevent
cross-alignment of chains,i.e. both chains of one complex aligning to a single chain of the other complex we
use a modified dynamic programing approach (See the COTH
server for details) in order to prevent cross-alignment.
SPRING uses a different approach for identifying protein complex templates. Structural analogs
of the individual constituents are identified by fold recognition programs suchas MUSTER or LOMETS. The monomeric templates are inputted into a pre-calculated
lookup table to identify protein complexes in the PDB that share an orientation similiar to the query complex. The lookup table is generated from a non redundantet
set of all pairwise oligimers from the PDB. SPRING selects and ranks complex analogs by a combination of monomer threading Z-scores, interface contact statistical potential, and the TM-align match between monomer-to-dimer templates.
The protein complex structure library is however largely incomplete. To remedy this problem, the TACOS server simultaneously runs
the LOMETS single chain threading for both the chains of the query complex. Since LOMETS is run on the much larger protein tertiary structure library the templates identified by LOMETS for the individual chains are of better quality than that identified by TACOS threading only. Following the generation of the LOMETS individual chain templates, therefore the TACOS server superimposes the LOMETS templates onto the corresponding chain of the multimeric template framework. This ensures a much improved quality of the individual chains while maintaining the orientation information identified by multimeric threading.
| How does TACOS use the templates to generate full-length structures? |
Once the templates have been identified, they are broken into fragments and placed on the CAS on-off lattice. The templates unaligned regions are then first build using a C-alpha random walk to generate initial full length structures. These full length dimers are then mapped onto an artificial monomer by using a completely flexible pseudobond to connect the C-terminal of the first chain with the N-terminal of the second chain. A figure showing an example of the lattice-based respresenation of the dimers is shown in Figure 1. The structures are then rigorously sampled using a number of intra-chain bond rebuilding and rigid body moves as well as a large inter-chain move for sampling the orientational space betweent the interacting protein subunits. To guide the sampling, a new force-field was designed comprising of knowledge-based energy terms derived from the protein complex structure library and template-based distance and contact restraints.
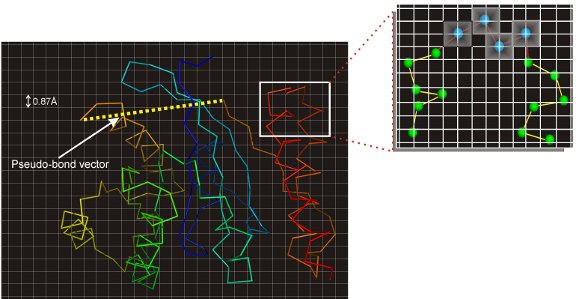
The decoys generated by the sampling scheme are then clustered by SPICKER and the cluster centroid of the largest clusters are identified. These cluster centers are then refined to remove clashes, generate full-atomic structures and to optimize the hydrogen-bonding network by FG-MD. These full-atmomic refined results are finally returned as the final models. For the complete flowchart of the TACOS algorithm please refer to Figure 2.
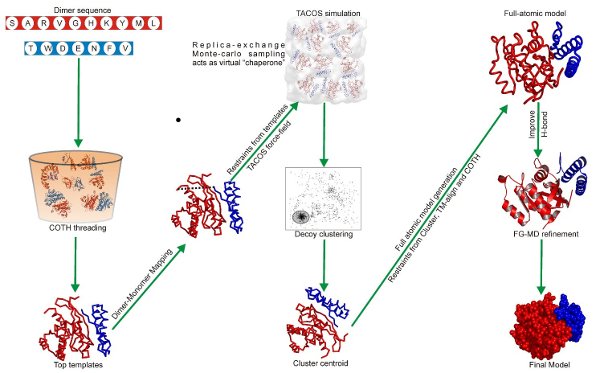
| How to cite TACOS |
You are requested to cite following articles when you use the TACOS server:
| Contact information |
The TACOS server is in active development with the goal to provide the reliable protein complex structure predictions. Please help us achieve the goal by sending your questions, feedback, and comments to: yangzhanglab@umich.edu.
|
yangzhanglab |